가장 많이 이용되는 항체 antibody database인 OAS (Observed Antibody Space)와 SabDab(The Structural Antibody Database)을 소개한다.
항원, 항체의 구조 분석은 연구에 가장 기본적으로 이용 되는 방법이다.
신약 개발에도 구조와 염기 서열을 바탕으로 여러 기능 연구를 하게 된다.
데이터 베이스 소개, PDB 아이디로 아미노산 서열 fasta 파일 다운받는법, 파이썬 코드를 통해서 데이터 베이스를 간단하게 정리하는 방법까지 함께 다루도록 하겠다.
연구에 가장 기본적으로 쓰이는 내용이고 면역학이나 바이오 인포나 학부 과제에 유용할 것이다.
항체 항원 데이터 베이스 ,OAS 와 SabDab
OAS 는 Observed Antibody Space. 지금까지 알려진 항체들을 모은 자료집이다. 직역하면 관찰된 항체 공간 이다.
OAS 중 단백질 구조가 있는 항체의 데이터 베이스는 SabDab이다. The Structural Antibody Database라고 한다.
이 데이터 베이스는 주기적으로 업데이트 되며, 연구에 필요한 정보들이 이미 정리되어 있다.
예를 들면,
- 데이터 수집 방법
- 결과를 발표한 논문 (링크) 및 저자 정보
- 유전자의 종 species - 사람인지 쥐인지, 라마인지 등등
- 만약 사람에게 받은 항체이면 그 사람이 이미 백신을 맞았는지, 어떤 질병을 가지고 있는지 (코로나 바이러스 항체 정보도 있다)
- 항체의 어떤 서열 (which chain : Heavy chain or Light chain etc) 인지.
- 항체 isotype
- B cell type (사람이 공여한거면, 공여자 나이)
- data-unit 에서 전체 염기 서열 갯수
- data-unit 에서 '유일한 염기서열' 갯수
등등 의 정보가 있다.
물론 OAS 의 데이터 베이스 종류가 하나가 아니라 약간의 차이는 있을 수 있다.
이렇게 자세한 주석과 정보가 있다는 점이 이 데이터 베이스의 가장 큰 장점이다.
This is well-organized and well- annotated database.
이 점은 논문에서도 강조되어 있다.
안티바디, 항체와 관련되어 다른 데이터 베이스 들도 많은데 - 돈 내고 다운 받아야 하는 상업 데이터 베이스도 있다- OAS와 SabDab는 연구자를 위해 무료로 잘 정리된 최신 정보를 제공하기 때문에, 많은 연구들이 이를 이용한다.
예를 들면,
- 논문에서 새로운 타겟을 찾을 때 스크리닝을 할 때 쓸 수 있다.
- 연구에서 구조가 없을 때 관련 항체나 항원 정보를 확인하고 모델링 하기 위해 쓸 수 있다.
- 머신 러닝이나 딥 러닝으로 알고리즘을 만들었을 때, 그 알고리즘의 효율을 확인하기 위해 쓸 수 있다.
참고로 oas의 전체 데이터는 700GB가 넘으니까 그냥 필요한 것만 다운 받는게 편하다.
OAS의 데이터를 이용할 경우에 관련된 논문을 인용해 주어야 한다.
아래는 인용정보이다. 혹시 데이터 베이스에 대해서 자세히 알고 싶다면, 논문을 무료로 볼 수 있으니 (open access) 참고하길 바란다.
OAS papers
Updated OAS paper: Olsen, T.H., Boyles, F., and Deane C.M. (2021). Protein Science.
The current OAS is an update of the previous paper: Kovaltsuk, A., Leem, J. et al (2018). J. Immunol.
SAbDab papers
Schneider, C., Raybould, M.I.J., Deane, C.M. (2022) SAbDab in the Age of Biotherapeutics: Updates including SAbDab-Nano, the Nanobody Structure Tracker. Nucleic Acids Res. 50(D1):D1368-D1372
Dunbar, J., Krawczyk, K. et al. (2014) SAbDab: the Structural Antibody Database. Nucleic Acids Res. 42(D1):D1140-D1146
OAS 홈페이지. More OPIG Resources를 누르면 다른 홈페이지 (예를 들면 SAbDab 홈페이지) 로 갈 수 있다.
OAS에서 항체 염기 서열 다운 받기
OAS 데이터는 wget 을 이용하는데, wget 이 없으면 conda install wget 으로 wget 설치하면 된다.
OAS에는 unpaired sequences 와 paired sequences 로 나누어져 있다.
| unpaired sequences | variable region의 heavy chain, light chain 이 함께 pair 짝지어지지 않은 것 | 약 1.5밀리언 (백만 오천) 서열 |
| paired sequences | variable region의 heavy chain, light chain 이 함께 있다. |
12만 서열 (121838 paired sequens) |
이 두 서열의 차이는 2021 년 논문에 잘 설명 되어있다.
항체는 heavy chain 과 light chain으로 구성 되어 있다.
그 전에 항체의 서열은 RNA 시퀀싱으로 이루어지는게 대다수 였는데, 이 방법으로는 각 RNA가 같은 항체에서 (같은 Bcell에서) 나온 건지 알 수가 없었다. 그래서 같은 항원으로 생긴 항체들이었어도, 하나의 항체에서 나온 서열인지는 몰랐던 것이다.
그런데 기술의 발달로, single cell RNA sequencing,즉, 세포 하나하나 따로 RNA 시퀀싱을 할 수 있게 되면서 단일 Bcell에서 생성되는 항체의 heavy chain 과 light chain 을 파악할 수 있어서 이 두 서열을 짝지을 수 (paired) 있게 된 것이다.
항체란게 나노바디나 모노바디가 아니면 heavy chian 과 light chain 이 함게 항원을 인식하게 된다.
즉, 같은 항원을 인식하는 항체여도 heavy chain 과 light chain을 뒤섞어 버리면 항원 인식을 하지 않을 수도 있는 것이다.
물론, unpaired sequences도 중요한 정보를 제공하지만, 위와 같은 이유로 paired sequences 정보는 연구에 귀중한 자료가 된다.
어쨌뜬 원하는 정보가 unpaired 인지 paired 인지 확인했으면 해당 홈페이지로 간다.
만약 어떻게 다운 받는지 궁금하면 각 메뉴의 HELP 를 보면 된다.
paired는 unpaired보다 데이터가 수가 적기 때문에 전체 데이터를 다운 받아도 (압축 파일기준) 193MB 면 된다.
OAS 웹 서버 -> Paired Sequences/ Unpaired Sequeces-> Search 에 가서 원하는 정보를 입력하고 (필터 적용 안 할 꺼면 *있음) 찾기를 Search 누른다.
아래는 paired sequence search results 이다.
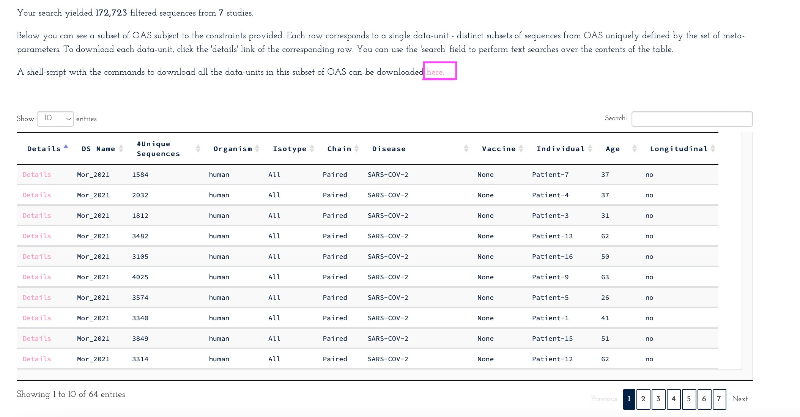
여기서 here 를 누르면 '정말 다운받을래?' 라는 문구가 뜬다.
다운 받으면 bulk_download.sh 파일이 받아진다.
wget http://opig.stats.ox.ac.uk/webapps/ngsdb/paired/Mor_2021/csv/SRR12875351_1_Paired_All.csv.gz
wget http://opig.stats.ox.ac.uk/webapps/ngsdb/paired/Mor_2021/csv/SRR12875354_1_Paired_All.csv.gz
wget http://opig.stats.ox.ac.uk/webapps/ngsdb/paired/Mor_2021/csv/SRR12875355_1_Paired_All.csv.gz
이런 식으로 된 wget 과 데이터 서버 위치가 있는 파일이다.
다운받는 방법은 해당 bulk_download.sh 파일을 내가 데이터 다운 받을 곳으로 옮겨 준다.
이제부터 터미널을 열어서 명령문을 써 주어야 한다.
윈도우면 파워쉘이나 모바엑스텀 같은 걸 이용해주면 같은 명령어로 쓸 수 있다.
mv bulk_download.sh 원하는 파일 위치.
그 다음에, 위 배쉬 파일을 실행가능하게 만들고, 실행시켜준다.

chmod u+rx bulk_download.sh
./bulk_download.sh
이제 다운 받은 데이터들은 파이썬 코드로 데이터프레임 만들어서 연구하면 된다.
다운 받은 파일들은 이미 aligned 되어 있다.
SAbDab에서 항체 구조와 염기서열 다운받기
OAS 와 SAbDab 는 살짝 다르다.
어떤 정보를 찾아야 데이터를 찾을 수 있는지 차이가 있다.
SAbDab는 구조가 있는 데이터 베이스라서
- 특정 서열에 맞는 구조 (또한 요정한 서열과 찾아낸 구조의 아미노산 서열 배열 paired-wise alignment)
- 특정 조건에 맞는 구조들 (구조 정보인 PDB id 등은 있지만 해당 PDB 의 항체 아미노산 서열은 없음)
위의 데이터에서는 특정 서열 조건 없이, 각 구조에 해당하는 아미노산이나 유전자 염기 서열이나, 여러 서열이 모두 정렬된 msa(multi-sequence alignment )정보를 얻기 쉽지 않았다.
thera-SabDab은 서열 제공, 총 827개의 치료용 항체들 정보가 있다.

원하는 구조를 찾으면 'summary' 데이터를 다운 받을 수 있다.
위의 OAS에서 wget을 이용한 것과 달리 그냥 csv/tsv 파일 다운 받으면 된다.
서머리 파일에 PDB - Hchain -Lchain -model .. 이렇게 정보가 있는데 한 열에 있는 Hchain 과 Lchain 이 'paired' 된 항체 정보이다.
1ahw B A 0
1ahw E D 0
이렇게 있으면,
PDB ID 1ahw 의 B와 A 체인이 하나의 항체를 이루고 ,B가 heavy chain, A가 light chain 이다.
PDB ID 1ahw 의 E와 D 체인이 하나의 항체를 이루고 ,E가 heavy chain, D가 light chain 이다.
0은 이 구조를 xray crystallography 즉 X선 회절 결정학으로 풀었다는 뜻이다.
Multi Sequence alignment 를 만드는 건 다음에 다루고, 일단,위의 데이터에서 PDB id를 통해 구조에 해당하는 아미노산 서열을 다운 받는 법을 소개해보겠다.
PDB ID로 아미노산 서열 fasta 파일 한번에 다운받기
일단 SabDab에서 원하는 조건들로 Get structures, 를 눌러서 서머리 페이지를 다운 받는다.
SabDab-> Structure Search -> Search Structures by attribute 에 오면 아래와 같이 조건을 지정할 수 있다.
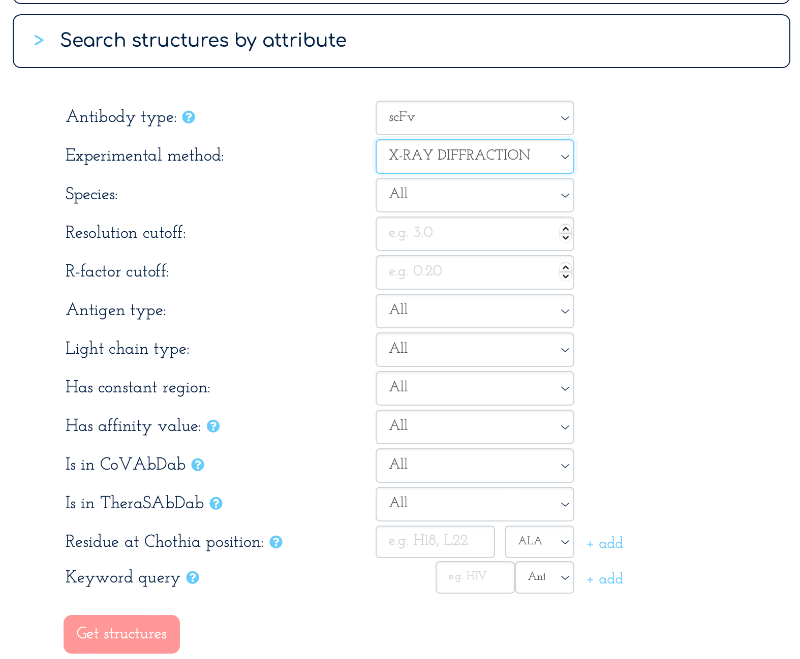
위의 조건에서는 161개의 구조가 발견되었다.
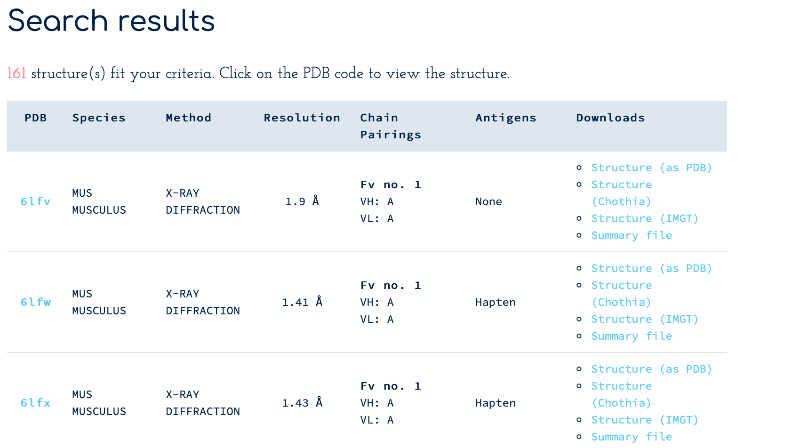
화면을 아래로 내리면 아래와 같은 다운로드 페이지가 보인다. 파란색 summary file을 다운 받는다.

이 서머리 파일은 엑셀로도 열 수 있다.
여기서 목표는 https://www.rcsb.org/downloads/fasta 에서 단백질 서열을 다운 받는 것이다.
우리는 단백질 파일의 다른 정보 말고 항체 정보만 얻고 싶기 때문에 아래와 같이 Asym IDs (Chain IDs)를 선택해준다.

이 선택에서는 PDB 는 모두 대문자여야 하고, 해당 체인과 온점으로 나뉘어 있어야 한다.
또한 각 체인은 쉼표 (,) 로 나뉘어 있어야 한다.
'1ahw B A 0 는 PDB ID 1ahw 의 B와 A 체인이 하나의 항체를 이루고 ,B가 heavy chain, A가 light chain이룬다.' 의 예시를 이용하면 다음과 같다.
만약, heavy chain, light chain 모두 다운 받고 싶다면,
1ahw 를 대문자로 바꾸어준 1AHW.B, 1AHW.A 를 위의 빈칸에 넣으면 되는 것이다.
방법 - 파이썬 코드
일단, 필자는 해당 파일 이름을 "SabDab_scFv.tsv" 로 바꾸었다.
편하게 쥬피터 노트북에서 보여드리겠다.
jupyter-notebook을 연다.
tsv 파일은 pandas 의 read_table 을 이용하면 된다.
그러면 아래 같이 표로 예쁘게 표현된 데이터베이스를 볼 수 있다.
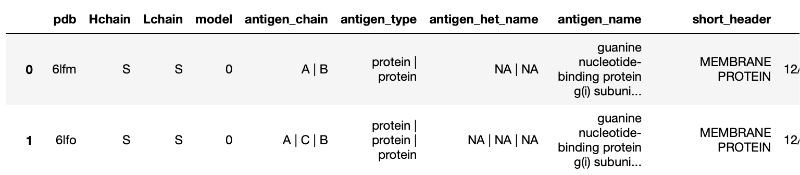
이제 PDB 코드를 대문자로 바꾸겠다.
이것 자체는 input_df['PDB']=[cur_pdb.upper() for cur_pdb in input_df['pdb']] 를 이용하면 된다.
- 파이썬 문자 String 에 .upper() 를 쓰면 모든 문자를 대문자로 바꾸어주는데 이걸 for loop 에서 이용한 것이다.
- pandas dataframe 판다스 데이터 프레임에서 새로운 열의 추가는 기존 열과 같은 길이의 리스트가 있을 때, 위와 같이
- 기존 데이터 프레이['새로운 열 이름'] = 기존 데이터 프래임과 같은 행 수를 가진 리스트 를 입력해준다.
아래와 같은 코드를 통해서 원하는 체인을 원하는 형식으로 출력한다.
본 코드는 글의 가독성을 위해서 쓸데없이 길게 썼다.
또한 이런 주석 방식도 선호되는 건 아니다.
그저 블로그를 위한 방식임.
위 코드는
- 일단 원하는 행의 정보가 NaN 이 아닌 것들만 추리고
- PDB 를 대문자로 바꾸어 주고
- PDB.체인번호 형식 즉, PDB Asym IDs 에 맞는 형식으로 적어주고 이걸 새로운 열 (out_col) 에 저장해주고
- 원한다면 필터를 이용해서 특정 행만 이용하고
- 각 PDB 당 1개의 chain 값만 텍스트파일로 저장해준다 (unique_id=False 이면 다 저장)
굉장히 간단한 건데.. 뭔가 말로 늘리고 그러다 보니 길어졌다.변수 이름등 보완할 점이 보이지만 일단 작동하는 코드이다.
아래는 그냥 랜덤한 정보들
- def ... return 이게 파이썬에서 함수 지정하는 법이다.
- def get_pdbchainid 에서 get_pdbchainid가 함수 이름이다.
- input_df, fname, col_name="Hchain"... 이 입력할 함수 정보 들이다.
- col_name=".. 처럼 = 으로 값이 지정된 것은 따로 정보를 안주면, 이미 지정된 값을 이용하겠다는 뜻이다. optional 옵션이다.
- 함수 코드를 만들 때 옵션은 필수 입력값 뒤에 와야 한다. 즉 ,get_pdbchainid(input_df,col_name="Hchain", fname, ) 이렇게 쓰면 옵션값인 col_name 이 필수 값이 fname 보다 앞에 와서 오류뜬다. 즉, 에러 난다.
- 위 코드에서는 필터를 적용하고 싶으면 하게 했다. 기본 값은 L 정보가 있으면 이용안 한다는 것이다. 그게 아니면 filter_col=[False,None] 적용 해준다.
- H,L 둘다 하고 싶으면 col_name 을 Lchain 로 하고 out_col PDB_Lchain 등으로 바꾸어서 filter_col=[False,None] 적용해서 get_pdbchainid 실행 해준다.
- 판다스에서 NaN 값 확인은 isna() 로 한다.
sabdab_scfv_df,_,_,PDB_Hchain = get_pdbchainid(sabdab_scfv_df,'sabdab_scfv_df_Honly_noLfilter',filter_col=[False,None])
이런식으로 이용하면 된다.
앞에 _,_ 는 출력값 저장 안할 꺼라 저렇게 해준 것이다.
어쨌든 이러면 이제 텍스트 파일 내용을 복사해서 붙여넣으면 fasta 파일이 다운받아진다.
오류가 될 수 있는 부분이 있었다. 최신 PDB에서 저자들이 입력한 체인 값과 다른 값을 지정했을 경우 (F [auth R]) 이런식으로 표현 된 경우에는 저자들이 입력한 값으로는 해당 fasta 값이 없다고 에러가 난다. SabDab 의 체인 값은 기본적으로 '저자들'이 제공한 값이 제공된다. 이건 PDB 가 새로 기능을 업데이트 하면서 생긴일 같다.
어쨌든 다운 받아진 파일은 아래와 같은 형식이다
>7RG9.E|nanobody 35|Lama glama (9844)
MKYLLPTAAAGLLLLAAQPAMAQVQLQESGGGLVQPGGSL...
즉 > PDB ID. Chain number | 어떤 체인인지 설명 | 어느 종에서 나온건지 설명.
아미노산 서열.
이런식으로 일정형식이 있으므로 파이썬 코드로 한줄 씩 읽어서, 해당 PDB 데이터 프레임에 아미노산 서열을 입력해 준다.
이는 아래 코드에 설명되어있다.
이제 이 정보들을 이용해서 sequence alignment 를 하거나. sequence motif 를 찾거나 seqlogo 등 이용하자.
아니면 아미노산 서열 만으로 어느 종에 해당되는지 찾을 수 있나 classification 코드를 짜거나 새로운 아미노산 서열을 만들어 보거나 등등 여러가지를 하면 된다.
마무리
이렇게 데이터 베이스를 소개하고 어떻게 아미노산 서열을 PDB에서 찾는지 간단한 코딩 예시와 함께 소개했다.
pyfamsa_align 방법도 표현할까 하다가. . 일단 더 좋은 align을 찾으면 추가 하기로!
이때 alignment 방법과 seqlogo 도 소개하면 될듯..

'info : 유용한 정보, 체험기' 카테고리의 다른 글
| 파이썬으로 데이터 입력 하기 및 삭제하기 pandas 를 이용해 csv 파일 편하게 수정하기. (0) | 2022.11.15 |
|---|---|
| 마크 다운 mark down 에서 글 중간에 글씨체 바꾸기 - font-family (0) | 2022.11.14 |
| 우울, 불안, 무기력 인가? 심리상담사 찾기, 가격, 학교 무료 상담, mental health coverage (0) | 2022.11.05 |
| 딥 러닝 GNN, Message Passing Neural Network 메세지 패싱 뉴럴 네트워크 (0) | 2022.11.02 |
| 여행 경비 정산 어플 추천, 경비 정리에 편한 어플 추천 + 미국 여행 필수 어플 추천, 여행 준비물 (0) | 2022.10.30 |




댓글